SINGLEEDGES.R
Description
This R script takes the output from the blast.pl script, a hash file containing query names and their respective chromosome numbers ( queryIndex) and a hash file containing hit names and their respective chromosome
numbers ( hitIndex). The script outputs either one PDF per query or per target (depending on the user's liking). Each PDF contains nodes for all queries and targets, but unlike the output from edges.R,
which is a many-to-many diagram, this script creates either a
one-to-many (all targets matched by a single query) or a many-to-one
(all queries which match a single target) diagram.
Arguments
- out.txt (Table with BLAST hit information)
- queryIndex (Hash of subject sequences)
- hitIndex (Hash of target sequences)
*These files must all be in the same directory as the R script.
- outDir (directory where the pdfs are saved. Initially set to "edges/", but can be changed by editing the top of the R script)
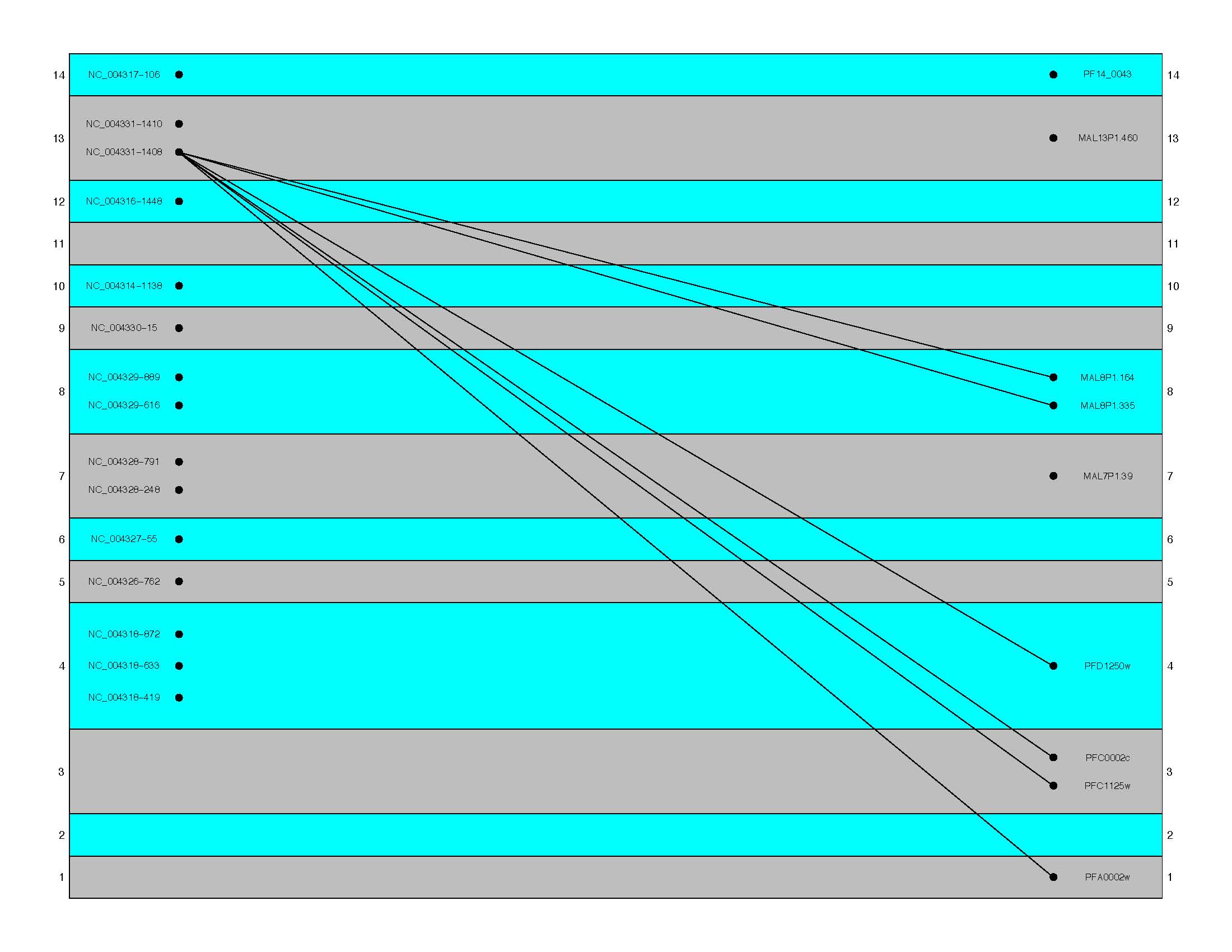
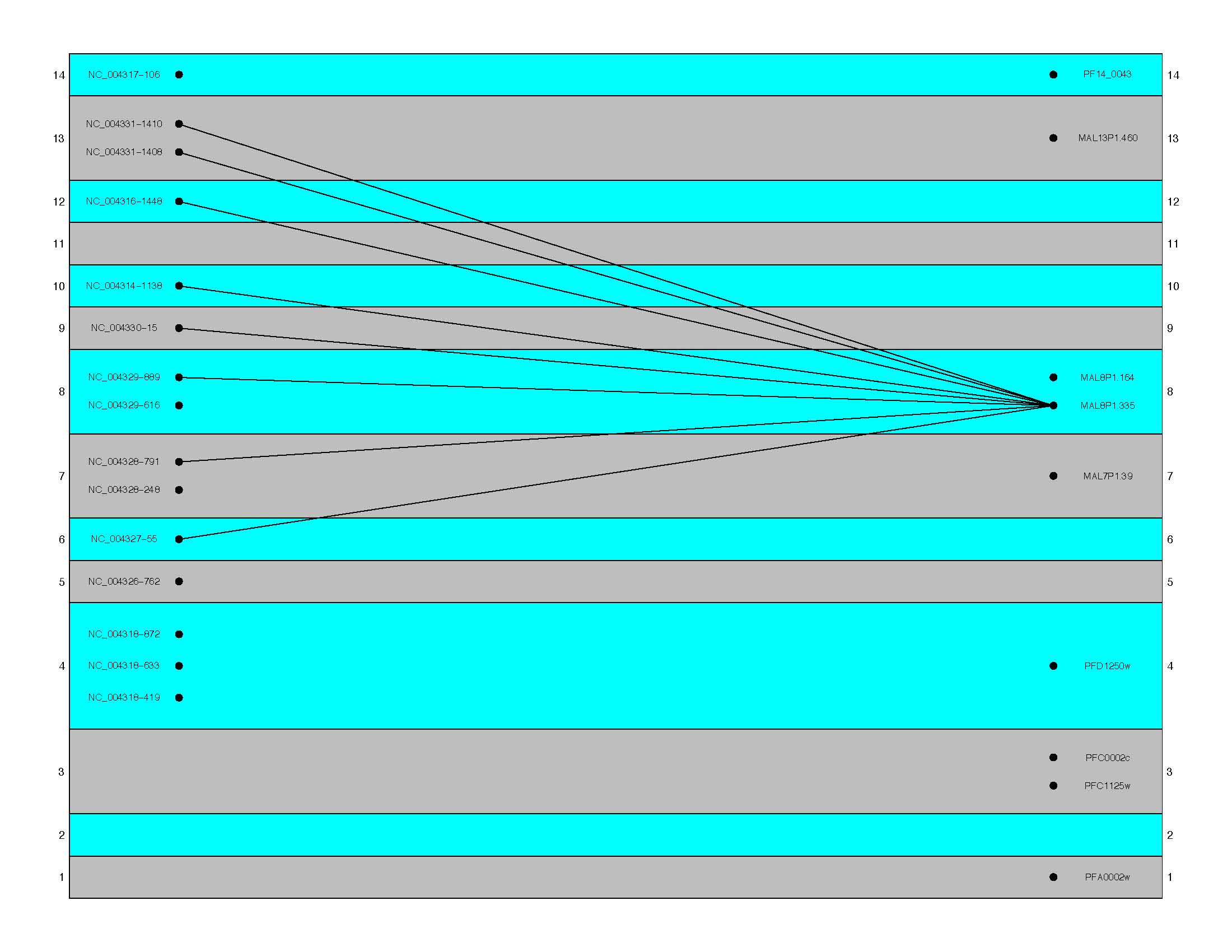
- oneQueryAllHits
(Default = TRUE. This produces results as seen in the first
figure below. When set to false, results are similar to those
seen in the second figure)
Notes
**IMPORTANT**
Before this script can be run, you must ensure that there is a sub-directory named "edges". The script creates PDF files
and places them there. If no such directory is created the
following error (or something similar) will be generated:
Error in pdf(paste(outDir, targetName, ".pdf", sep = ""), width = pageWidth, :
unable to start device pdf
Usage:
The R scripts are all made so that they can be sourced into R. From an R prompt type:
> source("singleEdges.R")
Output:
One PDF file per target sequence is created in the sub-directory "edges". Each filename has the structure SEQUENCENAME.pdf.
oneQueryAllHits = TRUE
 oneQueryAllHits = FALSE
oneQueryAllHits = FALSE
|