Description
ChIP-PED is designed to enhance the analysis of ChIP-chip and ChIP-seq (ChIPx) data. Given the target genes of a TF in one or more cell types, ChIP-PED searches for new biological systems potentially enriched with regulatory activity of the TF by superimposing ChIPx data on large amounts of Publicly available human and mouse gene Expression Data from a diverse collection of biological systems. This allows users to extend the scope of their ChIPx data to possibly novel biological systems without performing additional costly and time-consuming ChIPx experiments.
Requirements
Due to the large size of the gene expression compendium data sets, ChIP-PED requires the user to run the 64-bit vresion of R and have at least 4GB of available memory.
How to Install
First, download and install the 64-bit version of R from here.
Next, download both ChIPPED R packages using the links on the right side. ChIPPED is split into two packages - ChIPPED, which contains the main ChIPPED functions, and ChIPPEDdata, which contains the gene expression compendium data.
If you do not have the bigmemory package installed, please install it first:install.packages("bigmemory")
Then to install the ChIPPED and ChIPPEDdata packages, type in R:
install.packages(".../ChIPPEDdata_0.99.0.tar.gz", repos=NULL, type="source")install.packages(".../ChIPPED_0.99.2.tar.gz", repos=NULL, type="source")
where ... is the path that you saved the file.
Alternatively, if you are using linux, type in the command line of linux:
R CMD INSTALL .../ChIPPEDdata_0.99.0.tar.gzR CMD INSTALL .../ChIPPED_0.99.2.tar.gz
For more details on how to install R packages, see here.
How to Use
ChIP-PED requires as input the TF and list of activated (positive) and repressed (negative) target genes (TGs) in Entrez GeneID format. The activated and repressed target genes can be defined using ChIPx and corresponding gene expression data in which the TF expression is perturbed through knockout, knockdown, or etc. methods. Activated target genes are then genes that are both TF-bound in the ChIPx data and differentially increase in expression when the TF expression increases in the gene expression data. Similarly, repressed target genes are then genes that are TF-bound and differetially decreases in expression when the TF expression increases.
ChIP-PED also requires the user to define a regulatory pattern and region of interest. Four regulatory patterns of interest are possible: TF+TG+, TF+TG-, TF-TG+, and TF-TG-.TF+: ChIP-PED searches for samples with TF expression ABOVE the TF expression cutoff.
TF-: ChIP-PED searches for samples with TF expression BELOW the TF expression cutoff.
TG+: ChIP-PED searches for samples with TG activity ABOVE the TG activity cutoff.
TG-: ChIP-PED searches for samples with TG activity BELOW the TG activity cutoff.
Thus, TF+TG+ means ChIP-PED will search for samples with TF expression ABOVE the TF expression cutoff AND TG activity above the TG activity cutoff.
The region of interest and then set though the TF expression and TG activity cutoffs by three possible methods:
Norm: the TF expression and TG activity cutoffs will be set based on a fitted normal distribution, where the cutoffs correspond to a user inputted p-value (one-sided).
Quantile: the TF expression and TG activity cutoffs will be set by quantiles, where the cutoffs correspond to the user inputted quantile.
Exprs: the user will directly specify the TF expression and TG activity cutoff as input.
Thus, if the user selects Quantile, then they would also choose a quantile between 0-1. ChIP-PED will then set the TF expression and TG cutoff such that they correspond to the chosen quantile.
In addition, ChIP-PED offers users the option to perform automated follow-up analysis for each prediction (as explained in 'ChIPPED Further Exploration') using by setting Detail to TRUE and specifying a directory to save the results using directory. Each predicted context will be further anlayzed along with related contexts using tabSearch and ChIPPEDeda.
Example 1 ChIP-PED analysis
For example, if we were interested in studying the target genes of the transcription factor Oct4 using target genes derived from Oct4 ChIPx data in embryonic stem cells, we would type the following into R:
library(ChIPPED); library(ChIPPEDdata)
data(Oct4ESC_TG)
head(Oct4ESC_TG[[1]])
head(Oct4ESC_TG[[2]])
Output <-
ChIPPED(TFID="18999",PosTG=Oct4ESC_TG[[1]],NegTG=Oct4ESC_TG[[2]],
pattern="TF+TG+",coType="Norm",TF.co=0.1,TG.co=0.1,species="Mouse",Pval.co=0.05)
head(Output[[1]])
Next, we inputted the Entrez GeneIDs of the TF and its target genes into the main ChIPPED function, and also specified the regulatory pattern of interst as TF+TG+. For the region of interest, we specified the cutoff type (coType) as "Norm", meaning the TFco and TGco parameters, both inputted as 0.1, will set the TF expression and TG activity cutoffs such that they each correspond to one-sided pvalues of 0.1 based on fitted normal distributions.
Finally, we let ChIP-PED know to use the mouse gene expression compendium (GPL1261) by inputting the species and told it to report all biological contexts signfiicantly enriched at an adjusted p-value less than 0.05 using Pval.co.
Output is a list that will contain the ranked table of predicted biological contexts as the first item, where the top-ranked biological contexts are more likely to be enriched with the TF+TG+ regulatory pattern of Oct4.
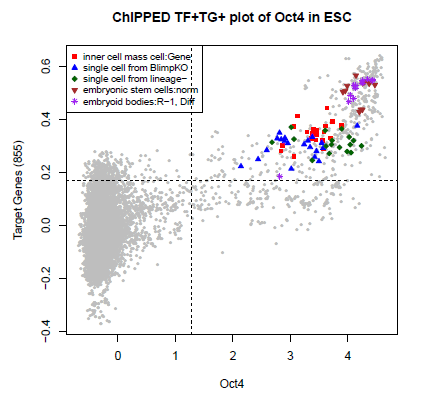
To visualize the output, we can use the ChIPPEDplot function to
generate a scatter plot of TG activity versus TF expression, where
the predicted biological contexts are highlighted in color:
ChIPPEDplot(Output,N=5,plotfile=NULL,Title="ChIPPED plot of Oct4 in ESCs", TFname="Oct4")
The resulting scatterplot will then look like:
ChIPPED Further Exploration
Sometimes users will be interested in a specific set of related biological contexts or would like to examine one of the resulting significant biological contexts in more detail. In order to do this, we provide two additional functions, tabSearch, and ChIPPEDeda, which together allows easy further exploration of the intial ChIPPED data analysis.For example, suppose we performed a ChIPPED analyis using STAT1 and its activated target genes defined from literature and HeLaS3 cells as below:
library(ChIPPED); library(ChIPPEDdata)
data(STAT_TG)
head(STAT_TG)
Output <- ChIPPED(TFID="6772",PosTG=STAT1_TG,NegTG=NULL, pattern="TF+TG+",coType="Norm",TF.co=0.1,TG.co=0.1,species="Human",Pval.co=0.05) head(Output[[1]])
Searchout <- tabSearch("GSE7123","Human")
Searchout
tabSearch(c("PBMC","hepatitis"),"Human","OR") tabSearch(c("PBMC","hepatitis"),"Human","AND")
ChIPPEDeda(TFID="6772",PosTG=STAT1_TG,NegTG=NULL,pattern="TF+TG+",coType="Norm", TF.co=0.1,TG.co=0.1,species="Human",Pval.co=0.5, SearchOutput=Searchout, Ordering="Average",Title="STAT1 GSE7123", plotfile=NULL,tabfile=NULL)
ChIPPEDeda will then generate four plots:
[A] A plot of mean +/- one standard deviation of the E_TF and A_TG values for each inputted context, where the order is set by the Ordering argument:
[B] A plot of TF expression (E_TF) versus TG activity (A_TG) across all samples in the compendium, similar to the ChIPPEDplot function, for the inputted contexts:
[C-D] Two heatmaps displaying the results (the t-statistics[C] and the p-values[D]) of t-tests comparing mean E_TF and A_TG values for each pairwise combination of inputted contexts. The top half of the heatmaps correspond to testing E_TF and the bottom half of the heatmaps correspond to testing A_TG.
We can see from [A] that healthy PBMCs have the lowest ETF and ATG values, while infected PBMCS especially those with shorter recovery durations after infection (day1 and day2) have the highest ETF and ATG values. From [B], we see more generally individual samples decrease in functional activity as more days are given for recovery, while healthy PBMCS have the lowest functional activity overall (black). From [C-D], we see that the t-tests comparing hepatitis-C infected PBMCs (day 7,14,28) have significantly increased ETF and ATG values compared to healthy PBMCs, and hepatitis-C infected PBMCs (day1,2) have significantly increased ETF and ATG values compared to hepatitis-C infected PBMCs (day7,14,28) and healthy PBMCs. Overall, the additional results allow us to show that STAT1 functional activity appears to be gradually decreasing as hepatitis-C infected PBMCs are given more time for recovery.
In addition, tables containing the data used to generate the plots will be reported along with the raw E_TF and A_TG values for further analysis.
We find that normal PBMCs cells fall outside of the TF+TG+ cutoffs, and are also not a significant STAT1 enriched biological context. This suggests that only during hepatitis C infection are PBMC cells enriched with STAT1 activity.
If we would like to save the plots, we can specify a file path in plotfile or if we would like to save the result tables, we can specify a file path in tabfile, otherwise the plots and tables will be directly reported in the R terminal. The file paths need to be ***.pdf for the plotfile and ***.csv for the tabfile.
If users would like to automatically perform these additional follow-up analyses after their initial ChIPPED analysis, they can use the ChIPPED function and then set Detail=TRUE and provide a directory to output the files:
Output <- ChIPPED(TFID="6772",PosTG=STAT1_TG,NegTG=NULL, pattern="TF+TG+",coType="Norm",TF.co=0.1,TG.co=0.1,species="Human",Pval.co=0.05, Detail=TRUE,directory="~/STAT1output")
For more details on how to use the tabSearch and ChIPPEDeda functions, please see the accompanying vignette and the R help files in the ChIPPED package.
Example 2 Target Gene Construction
In order to use ChIP-PED, users are required to construct their own target genes from ChIPx data and TF perturbation data. Thus, they will need to define the activated TF target genes and the repressed TF target genes, where activated targets are TF bound and positively differentially expressed and repressed targets are TF bound and negatively differentially expressed.
ChIPx peak detection
To begin, the first step is to detect significant peaks from your ChIPx data. One option is to use CisGenome. Afterwards, you should have a file that contains the significant peaks where the first column is the chromosome, the second column is the start of the peak, and the third column is the end of the peak, and additional columns can be other related, but not required info from the peak dection output. Here is an example output using ChIPx data for Oct4 in ESCs from GSE11491: GSM288346_ESC_Oct4.txt
Annotate ChIPx peaks
Next, we will need to annotate the significant ChIPx peaks to identify the TF-bound gene targets. To do so, we provide a R function in the ChIPPED package: annotatePeaks . annotatePeaks takes as input the output from the ChIPx peak detection, the genome to annotate the peaks with, and the annotation window region (upstream to downstream region of the TSS). For example, for the ESC peak deteciton output frmo above, we can first read-in the file then annotate all peaks with genes that overlap with the 10kb to 5kb region around the TSS of ecah gene from the mm8 genome:
path <- system.file("extdata",package="ChIPPED")
inputfile <- read.delim(paste(path,"GSM288346_ES_Oct4.txt",sep="/"),
header=FALSE,stringsAsFactors=FALSE)
annon.out <- annotatePeaks(inputfile,"mm8",10000,5000)
head(annon.out)
Microarray differential expression
The next step is to analyze your gene expression data from TF perturbation experiments to identify differentially expressed genes. We recommend using the work flows provided for R using Bioconductor packages: for example, one can preprocesse and normalize their gene expression data with RMA and then analyze the output for differentially expressed genes with limma . Be sure to also annotate the first column of the limma output with the EntrezGene ID corresponding to each probe. Here is an example output using gene expression data for Oct4 in ESCs, in which Oct4 is knocked-down via siRNA from GSE4189: Oct4_E14TG2a_GSE4189_Limma.txt.
Construct TF target genes
After analyzing both the ChIPx and TF perturbation data, then we only need to combine the two analyses by identifying the TF-bound and differentially expressed genes and whether they increase (activated) or decrease (repressed) in expression.
gp.out <- read.delim(paste(path,"Pou5f1_E14TG2a_GSE4189_Limma.txt",sep="/"),
stringsAsFactors=FALSE)
Oct4ESC_TG <- ConstructTG(annon.out,gp.out)
head(Oct4ESC_TG[[1]]) ## Activated (+) target genes
head(Oct4ESC_TG[[2]]) ## Repressed (-) target genes
ChIP-PED Analysis
Now, we are ready to perform a ChIP-PED analysis. Using the defined TF target genes and providing additional ChIP-PED parameters, we can obtain the final ranked table of significantly enriched Oct4-active biological contexts.
library(ChIPPED)
Output <-
ChIPPED(TFID="18999",PosTG=Oct4ESC_TG[[1]],NegTG=Oct4ESC_TG[[2]],
pattern="TF+TG+",coType="Norm",TF.co=0.1,TG.co=0.1,species="Mouse",Pval.co=0.05)
head(Output[[1]])
For more details on how to use the annotatePeaks and ConstructTG
functions and for how to perform more ChIP-PED analyses, please see the accompanying vignette and the R help files in the ChIPPED package.